 手機版
手機版 化工儀器網手機版
化工儀器網手機版
 化工儀器網小程序
化工儀器網小程序
 官方微信
官方微信 公眾號:chem17
公眾號:chem17
 掃碼關注視頻號
掃碼關注視頻號











藥物篩選在PROTAC中的應用_MedChemExpress(MCE 中國)

積極推進校企合作,整合多方優勢,協同創新平臺,解決新藥研發中的關鍵性技術瓶頸,助力生物醫藥產業生態共建!
近日,MCE (MedChemExpress) 與華東師范大學正式簽署合作協議,共同建立“AI 藥物探索聯合實驗室”,該實驗室將依托校企雙方的科研優勢與產業資源,重點突破 AI 技術在藥物研發領域的創新應用,構筑“AI +生物醫藥”創新高地,為新藥研發及科研成果轉化注入強勁動力。

作為世界前沿的科研化學品和生物活性化合物供應商,MCE 設有專業的實驗中心和嚴格的質控驗證體系,提供 HNMR、LC/MS、HPLC、手性分析、元素分析、SDS-PAGE、SEC-HPLC、活性檢測等各項質檢報告。MCE 深得全球數十萬科學家信賴,文獻引用高達 50,000+,產品種類豐富,涵蓋 80,000 多種特異性抑制劑,11,000+ 重組蛋白、3,000+ 染料、數百款試劑盒、抗體等,擁有為全球科研客戶及新藥研發群體提供一站式藥物發現的平臺,且不斷升級更新。
同時,MCE 多年的發展歷程和豐富的行業經驗,吸引了眾多資深的化學家和生物學家的加入,從產品的 HNMR 數據解析到生物活性數據,從客戶的詢價到產品的售后服務,MCE 擁有著完善的管理體系,服務更加高效、準確,得到了全球客戶的一致認可和好評。
我們始終將創新作為核心驅動力,積極探尋前沿科技在藥物研發中的應用,加速賦能全球超 13,000家合作伙伴實現從臨床前到商業化生產的全過程。MCE 愿積極承擔更多的責任和使命,深層次開展多領域合作,努力在藥物研發等領域取得重大的成果,為生物醫藥的研發創新做出更多貢獻!
華東師范大學作為國家A 類建設高校,在人工智能、計算化學以及藥物設計等多個學科領域擁有深厚的學術底蘊和強大的科研實力。此次攜手打造“AI 藥物探索聯合實驗室”,將整合雙方優勢資源,聚焦藥物空間探索應用場景,運用先進人工智能算法與大數據分析技術,著力解決新藥研發中的關鍵性技術瓶頸,為生物醫藥研發和科研成果轉化進程按下“快進鍵”。

傳統小分子藥物開發具有周期長,投入高,成功率低……隨著大數據技術的不斷進步,AI(人工智能)相關技術將在藥物設計中扮演更加重要的角色。AI 不僅可以提高藥物發現的速度,還能夠根據復雜的生物數據和化學信息進行智能分析,從而實現更為精準的藥物開發。
MCE 一站式藥篩平臺聚焦于藥物發現早期,積極擁抱 AI 帶來的巨大機遇和挑戰,將 AI 技術應用到藥物篩選、化合物庫設計等各個業務模塊,開發了多種 AI 算法助力藥物篩選服務。MCE 還通過 AI 算法生成 MegaUni 1,000 萬虛擬類藥多樣庫及 Mini 化合物庫的構建,幫助客戶更高效地獲得符合自己需求的化合物。
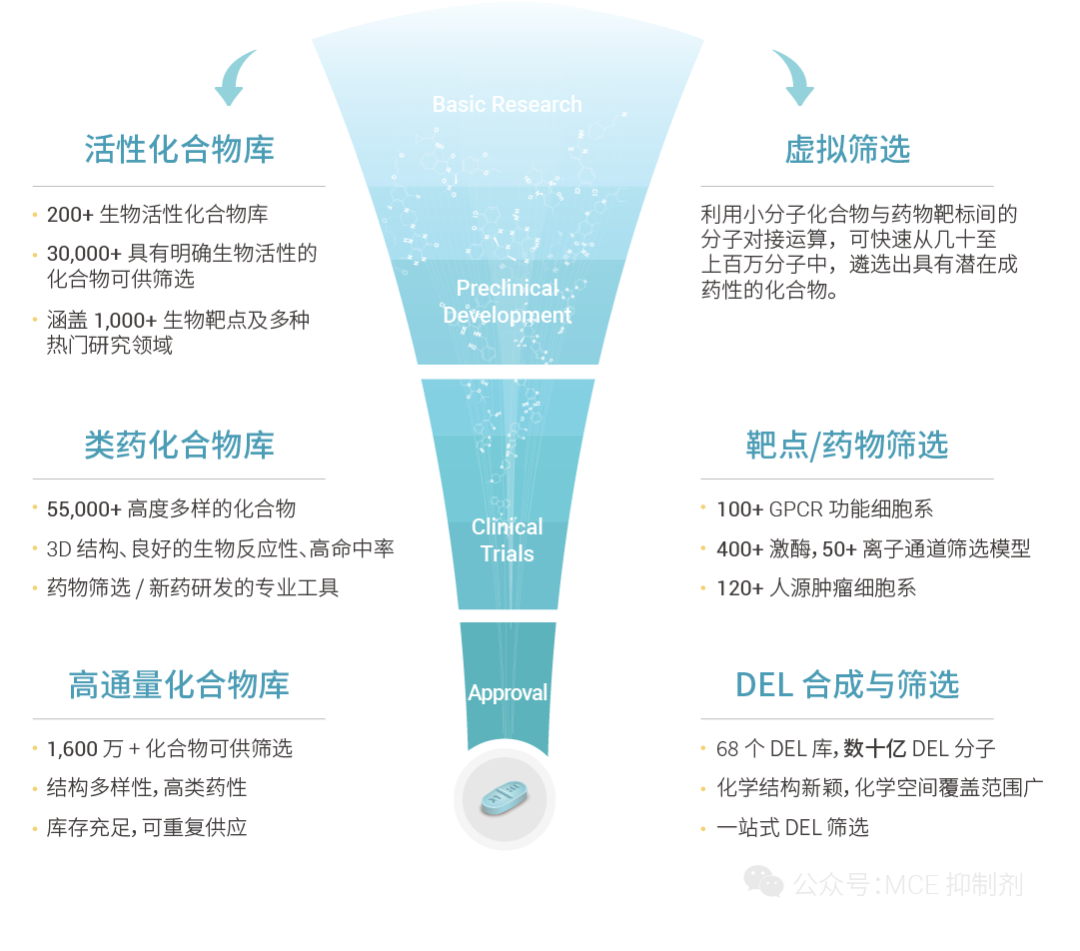
圖 1. MCE 一站式藥篩平臺簡介。
借勢咱們就來聊聊計算機輔助藥物設計 (CADD),當計算機遇到 PROTAC……是否能在一定程度上縮短 PROTAC 藥物開發的周期,提高成功率呢?本期看點:虛擬篩選、分子對接及分子動力學模擬技術在 PROTAC 開發中的應用!
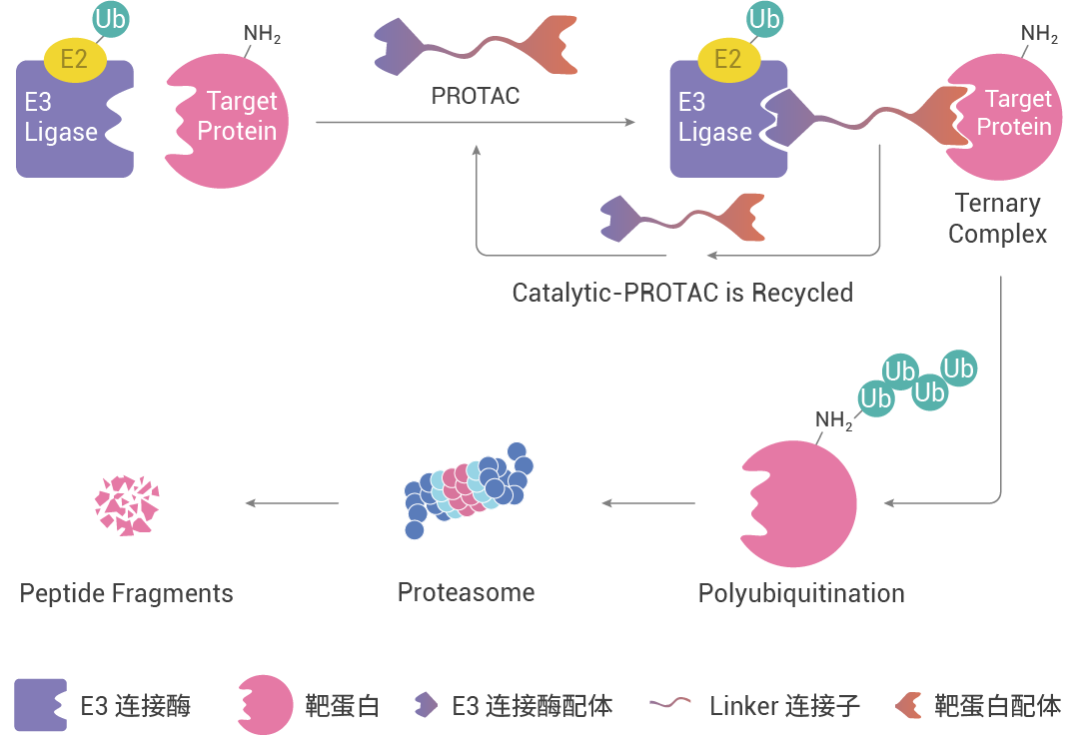
計算機輔助藥物設計 (CADD):
Section.01
虛擬篩選
在 PROTAC 設計中的應用
虛擬篩選是通過計算機模擬和預測的方法,在大量化合物數據庫中篩選出具有潛在結合活性的化合物,能夠大大加速藥物研發過程。靶蛋白配體(Ligand for Target Protein for PROTAC)作為 PROTAC 的導向基團,負責對靶蛋白的捕獲,因而需要有較高的配位能力與選擇性。
虛擬篩選技術在靶蛋白配體及 E3 泛素連接酶配體篩選中發揮重要作用。可以說虛擬篩選技術擴展了 PROTAC 技術的應用。
靶蛋白配體的篩選是 PROTAC 設計的第一步。靶蛋白的結合位點通常決定了配體的親和力和特異性。因此,了解靶蛋白的三維結構是進行虛擬篩選的前提。目前報道的 PROTAC 所用的靶蛋白配體多是已經報道的該靶蛋白的抑制劑,激動劑等。針對目前沒有相應配體報道的靶蛋白,虛擬篩選技術結合后期的實體驗證實驗可以加速靶蛋白配體的篩選,降低篩選成本。
近日,中國醫學科學院藥物研究所劉曉輝課題組就利用了虛擬篩選和體內外實驗成功獲得了親和分子#129,并進一步設計合成了一系列 PROTAC 分子。結果顯示,PT-129 能夠特異性地靶向降解 G3BP1/2,抑制應激細胞中應激顆粒的形成,并有效解體已存在的應激顆粒。后續的藥理學研究表明,PT-129 可以通過促使應激顆粒解體,阻斷 ATF4 在成纖維細胞與腫瘤細胞間的傳遞,顯著抑制了腫瘤細胞的增殖和體內腫瘤的生長[1]。
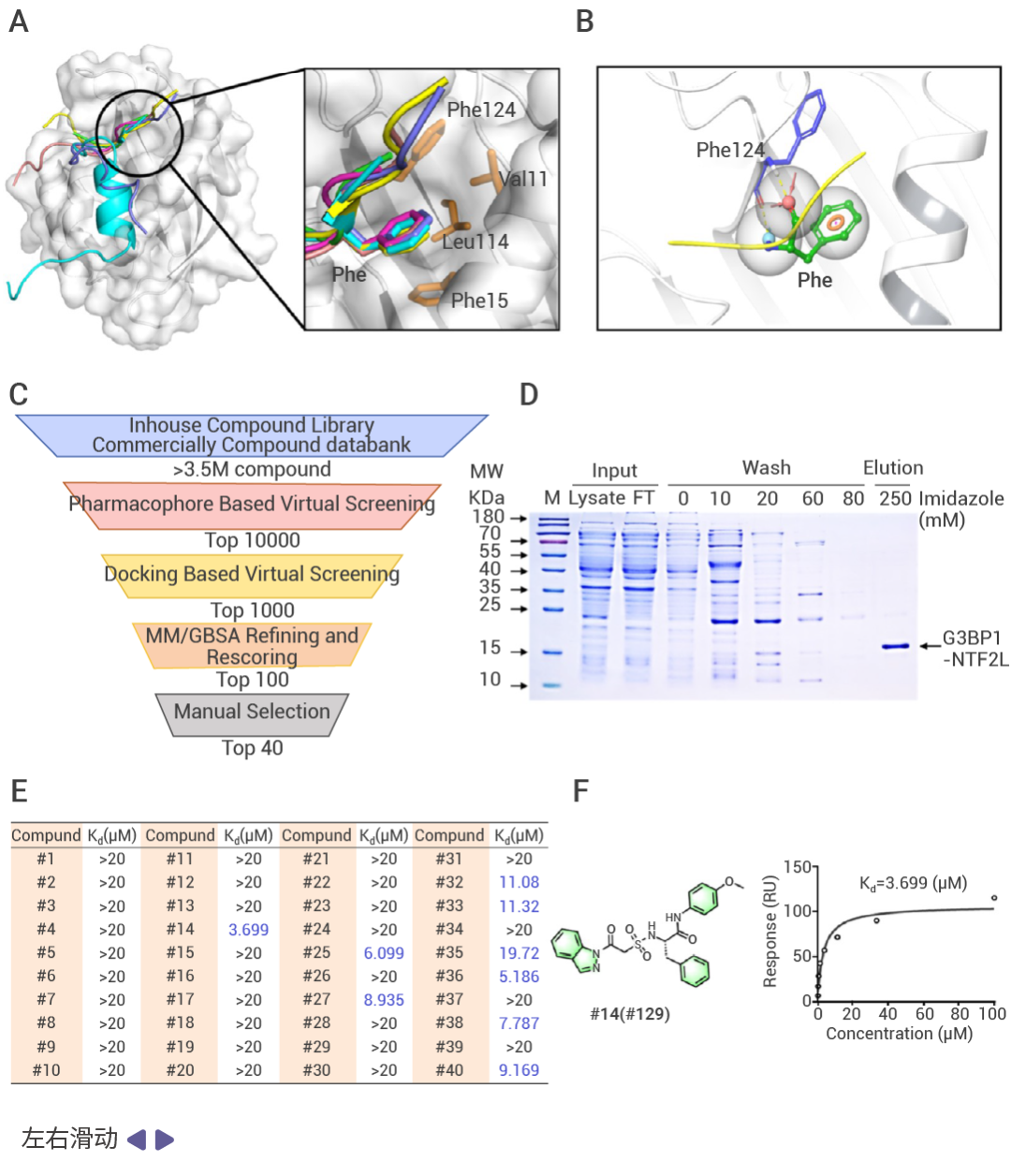
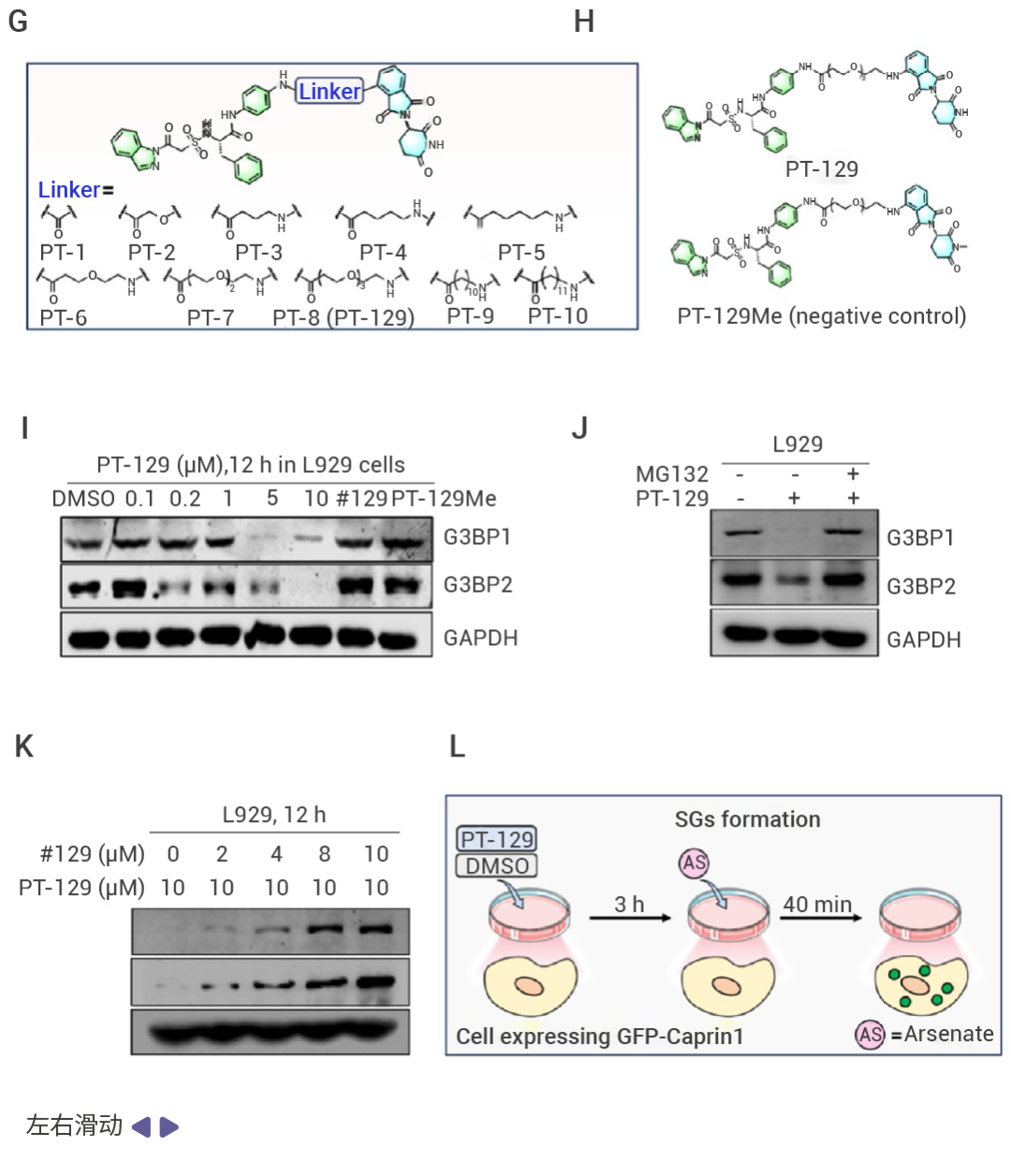
圖 2. 基于虛擬篩選和 SPR 的 G3BP1 蛋白配體篩選及 PROTAC 設計[1]。
Section.02
分子對接
在 PROTAC 設計中的應用
分子對接是分子模擬的重要手段之一,它依據小分子配體與蛋白受體作用的“鎖-鑰原理”,通過能量匹配、空間匹配和化學性質匹配而相互識別形成分子復合物,是模擬配體小分子與受體生物大分子相互作用的一種技術方法。
配體與受體相互作用是分子識別的過程,主要包括靜電作用、氫鍵作用、疏水作用、范德華作用等。通過分子對接預測靶蛋白和配體分子間的結合模式和親和力,也在 PROTAC 設計中發揮著重要作用。
在 PROTAC 設計中,Linker 與靶蛋白配體的附著位點從根本上影響 PROTAC 降解活性和選擇性,當配體與靶蛋白的共晶結構可用時,從這些高分辨率共晶結構中識別出配體的溶劑暴露位置可以確定為 Linker 附著位點。因此,可以通過分子對接或分子動力學模擬技術分析配體與靶蛋白結合關系來確定合適的 Linker 附著點。
Yalin Tu 等科學家在基于 EZH2 抑制劑 EPZ-6438 設計 EZH2 PROTAC 時,通過分子對接分析 EPZ-6438 的嗎啉環處于溶劑暴露區,可以作為 Linker 附著位點[2],所以在后續設計中,用 PEG Linker 替換嗎啉環,進行后續 PROTAC 設計,并獲得兩種有效的EZH2 PROTAC 分子,YM281 和 YM181。
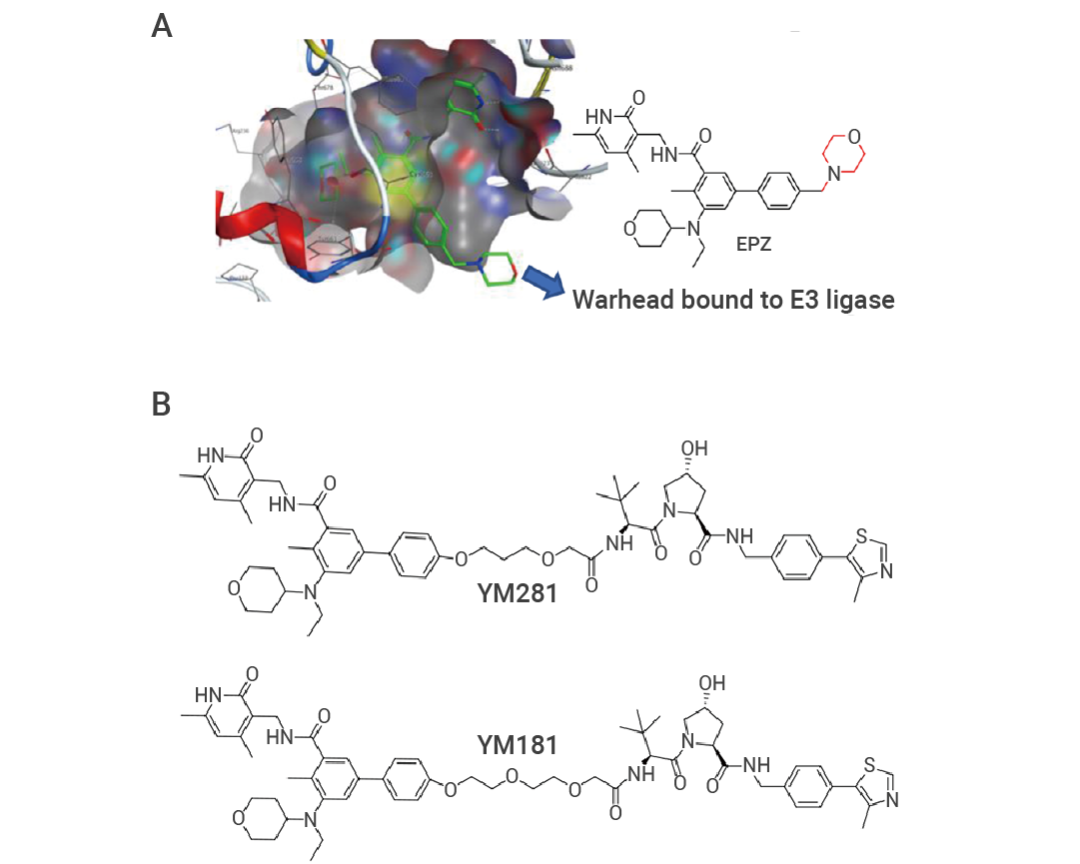
圖 3. EPZ6438 與 EZH2 活性口袋對接圖[2]。
Gilberto P. Pereira 等科學家基于 LightDock 工具開發了一種高效預測與 PROTAC 兼容的蛋白-蛋白相互作用界面的方法,在對包含 13 個三元復合物晶體結構的手動收集數據集進行基準測試時,該方法從結合結構出發的準確率達到 92%,從未結合結構出發的準確率達到 77%。該方法僅需提供 E3 連接酶和靶蛋白的單體形式的配體結合結構,因而具有通用性、準確性和高效性[3]。
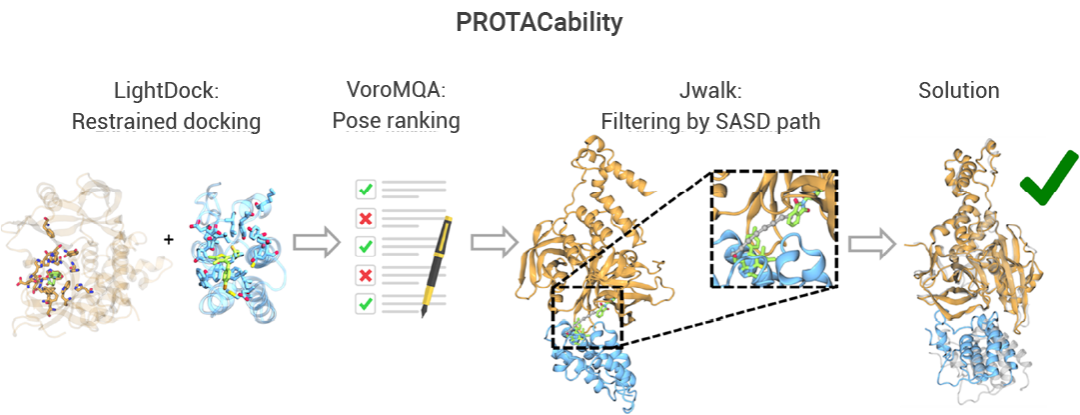
圖 4. LightDock PROTAC 對接流程[3]。
Section.03
分子動力學模擬
在 PROTAC 設計中的應用
分子動力學(Molecular Dynamics, MD)模擬是一種基于牛頓力學,綜合了物理、數學和化學等多種學科的計算機模擬方法,可以依據當前分子體系的位置、速度和動能等信息,推測該體系未來的位置、速度和動能,從而揭示分子運動的客觀規律。通過計算機分子模擬,研究人員能夠在分子水平上理解生物大分子的運動與生物功能、蛋白-小分子之間相互作用機理。
PROTACs 的核心要素之一,便是要擁有細胞膜通透性,確保它們能夠順利抵達目標蛋白所在之處。然而,這一要求在 PROTAC 的設計過程中構成了不小的挑戰。分子動力學模擬這一強大的計算機模擬技術,能夠深入探索分子的動態行為。通過 MD 模擬,可以精確分析 PROTAC 分子在細胞滲透方面的潛力,從而在 PROTAC 分子的初步設計階段就賦予其顯著的優勢。
Vasanthanathan Poongavanam 等人就使用了核磁共振光譜和分子動力學模擬,深入了解三種具有相似結構的靈活小分子 cereblon PROTACs,分析其在細胞滲透性方面表現出差異的原因[5]。研究表明,在非極性環境中,PROTACs更傾向于形成低溶劑可接觸的折疊構象,這一特性與它們的高細胞滲透性密切相關。鏈接子的化學性質和靈活性對于 PROTACs 聚集由分子內氫鍵、π-π 相互作用和范德華相互作用穩定的折疊構象是至關重要的。
通過此研究,研究人員得出結論,分子動力學模擬可用于在 cereblon PROTACs 的設計中前瞻性地評估細胞滲透性的排名。
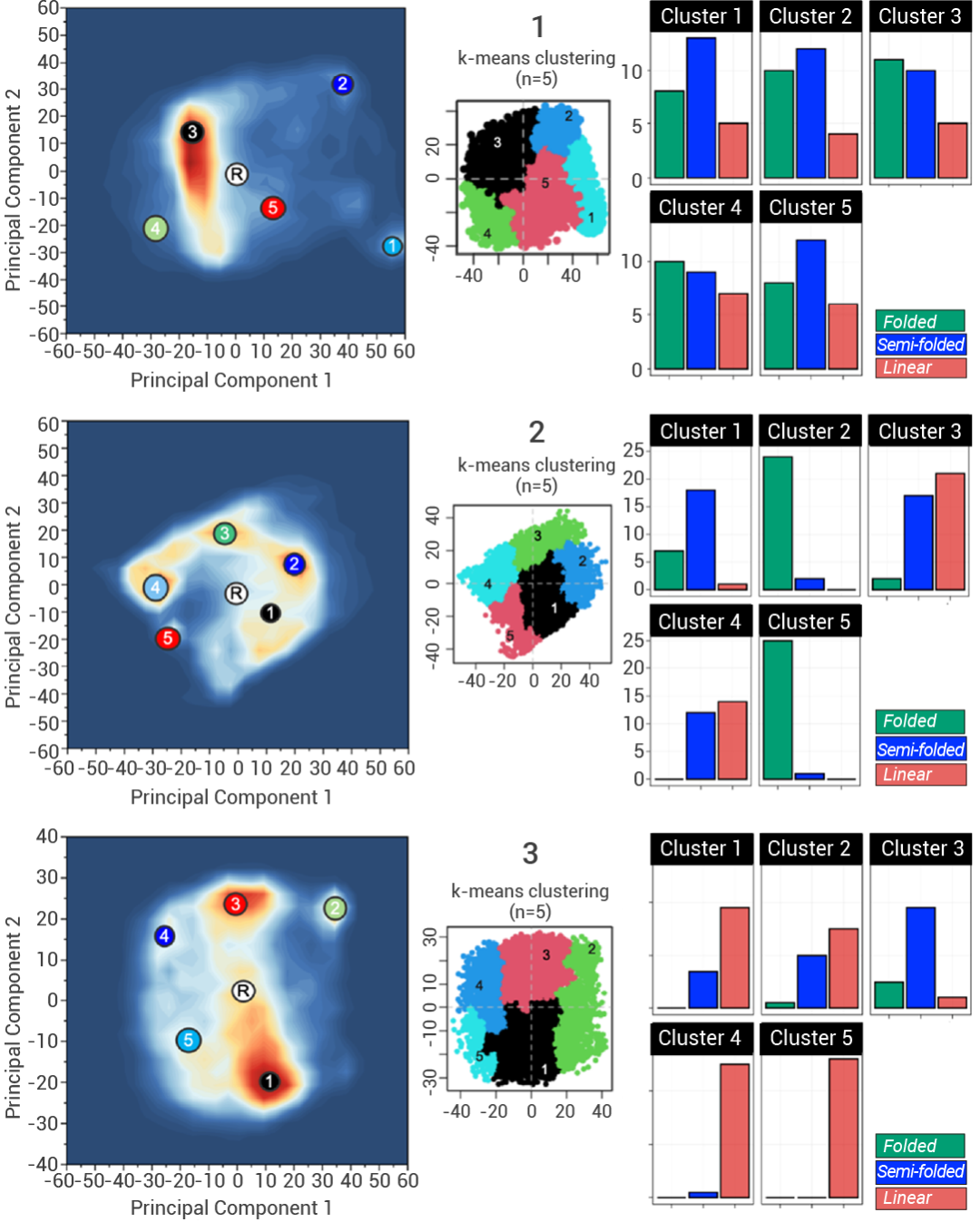
圖 5. 對三種 PROTAC 的分子動力學模擬[4]。
目前研究人員已經開發了幾種 MD 方法來模擬三元結構,深入理解 PROTAC 的機制,從而促進新型 PROTAC 的合理設計。
Section.04
小結
PROTACs 藥物的問世,標志著藥物研發領域的一場革命,盡管其發展歷程僅有短短二十年,卻已展現出巨大的潛力與希望。然而, PROTACs 藥物的開發仍面臨著諸多挑戰:當前,PROTACs 的研發大多依賴于一種“試錯法”——即通過不斷地調整 linker(連接子)的長度、成分、附著位點,或是更換配體類型,以期在無數次的嘗試中偶然發現一種具有活性的分子。相信隨著計算機技術及人工智能技術的不斷發展,CADD 及 AIDD 相關技術一定會在藥物設計領域有著更大的發展!
MCE 是世界前沿的科研化學品和生物活性化合物供應商,可以為科學家提供相關模塊化產品,包括熱門 E3 泛素連接酶配體、靶蛋白配體、Linker 及配體與 Linker 偶聯物,已報道的活性 PROTAC、SNIPER 等。同時,我們還提供 PROTAC 產品的一體化合成服務。
產品推薦 |
PROTAC 相關定制服務 PROTAC 全稱為 PROteolysis-TArgeting Chimeras (蛋白降解靶向嵌合分子),是一種雜合雙功能小分子化合物,結構中含有兩種不同配體,一個是泛素連接酶 E3 配體,另一個是與細胞中目標靶蛋白結合的配體,兩個配體之間通過 linker 相連,從而形成 “ 三體 ” 聚合物——靶蛋白配體-Linker-E3 配體。 |
虛擬篩選 虛擬篩選 (Virtual Screening, VS) 是基于小分子數據庫開展的活性化合物篩選。利用小分子化合物與藥物靶標間的分子對接運算,虛擬篩選可快速從幾十至上百萬分子中,遴選出具有成藥性的活性化合物,大大降低實驗篩選化合物數量,縮短研究周期,降低藥物研發的成本。 |
AI 篩選 人工智能 (Artificial Intelligence,AI) 藥物篩選是一種結合 AI 技術與計算化學的高通量篩選方法,廣泛應用于蛋白結構預測、新藥研發和分子設計與優化等領域。其主要目的是利用機器學習 (Machine Learning,ML) 算法分析大量數據,從中學習規律,生成 AI 打分函數,以此提高篩選效率,加速候選藥物的發現過程。 |
分子動力學模擬 分子動力學 (Molecular Dynamics, MD) 模擬是一種基于牛頓力學,綜合了物理、數學和化學等多種學科的計算機模擬方法,用于研究分子體系的運動和相互作用、預測分子體系的行為和結構性質。通過計算機分子模擬,研究人員能夠在分子水平上理解生物大分子的運動與生物功能、蛋白-小分子之間相互作用機理。 |

[1] Dong T, et al. G3BP1/2-Targeting PROTAC Disrupts Stress Granules Dependent ATF4 Migracytosis as Cancer Therapy. J Am Chem Soc. 2025 Jan 8;147(1):446-461.
[2] Tu Y, et al. Design, Synthesis, and Evaluation of VHL-Based EZH2 Degraders to Enhance Therapeutic Activity against Lymphoma. J Med Chem. 2021 Jul 22;64(14):10167-10184.
[3] Pereira GP, et al. Rational Prediction of PROTAC-Compatible Protein-Protein Interfaces by Molecular Docking. J Chem Inf Model. 2023 Nov 13;63(21):6823-6833.
[4] Poongavanam V, et al. Linker-Dependent Folding Rationalizes PROTAC Cell Permeability. J Med Chem. 2022 Oct 13;65(19):13029-13040.


免責聲明
- 凡本網注明“來源:化工儀器網”的所有作品,均為浙江興旺寶明通網絡有限公司-化工儀器網合法擁有版權或有權使用的作品,未經本網授權不得轉載、摘編或利用其它方式使用上述作品。已經本網授權使用作品的,應在授權范圍內使用,并注明“來源:化工儀器網”。違反上述聲明者,本網將追究其相關法律責任。
- 本網轉載并注明自其他來源(非化工儀器網)的作品,目的在于傳遞更多信息,并不代表本網贊同其觀點和對其真實性負責,不承擔此類作品侵權行為的直接責任及連帶責任。其他媒體、網站或個人從本網轉載時,必須保留本網注明的作品第一來源,并自負版權等法律責任。
- 如涉及作品內容、版權等問題,請在作品發表之日起一周內與本網聯系,否則視為放棄相關權利。


 采購中心
采購中心